


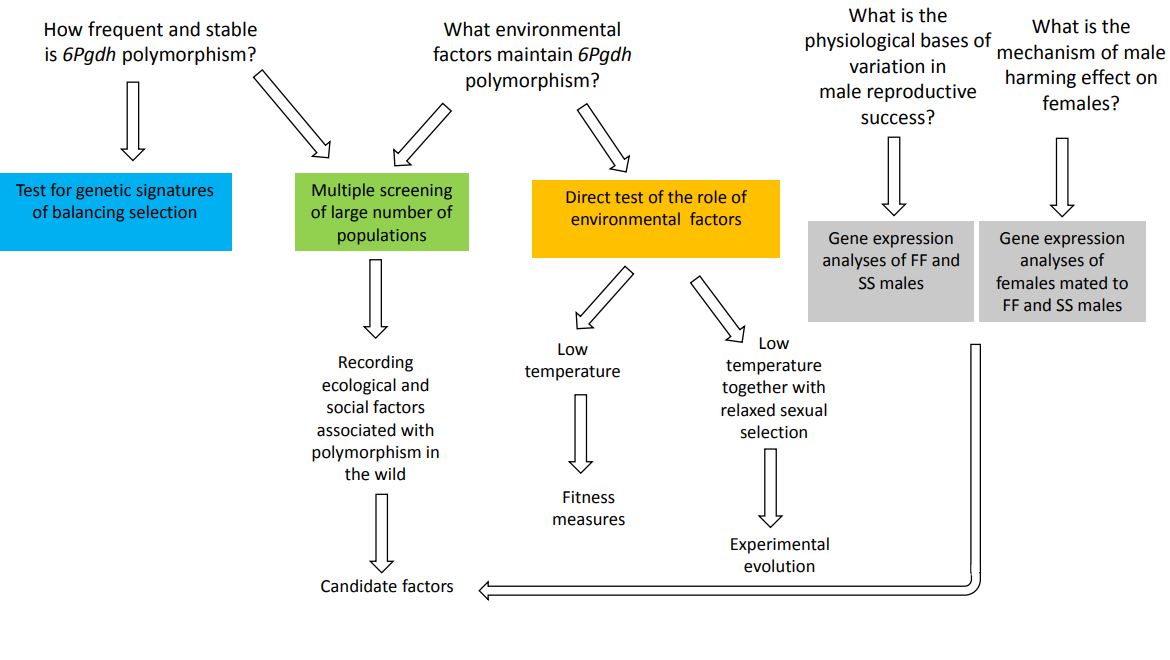
Environment-dependent balancing selection in a sexually selected gene

The ecology of ponds in the context of human activity and geography - environmental DNA and beyond


The role of blood parasites in emerging disease dynamics and biodiversity loss in amphibians

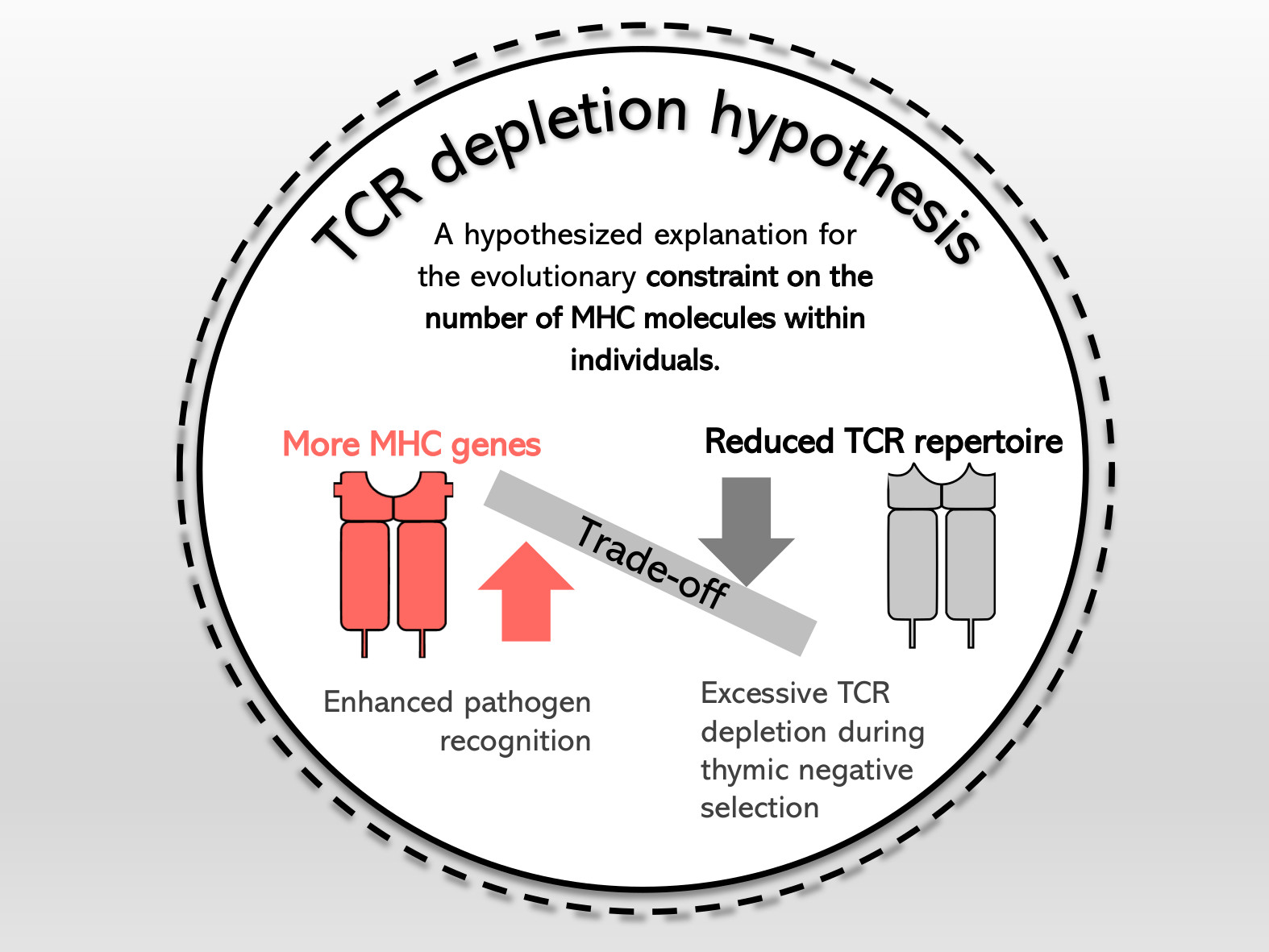
The evolution of non-classical MHC class I genes and T-cell receptor repertoires in salamanders