


Genomika gradacji - ewolucja neutralna i adaptacyjna u kornika drukarza.
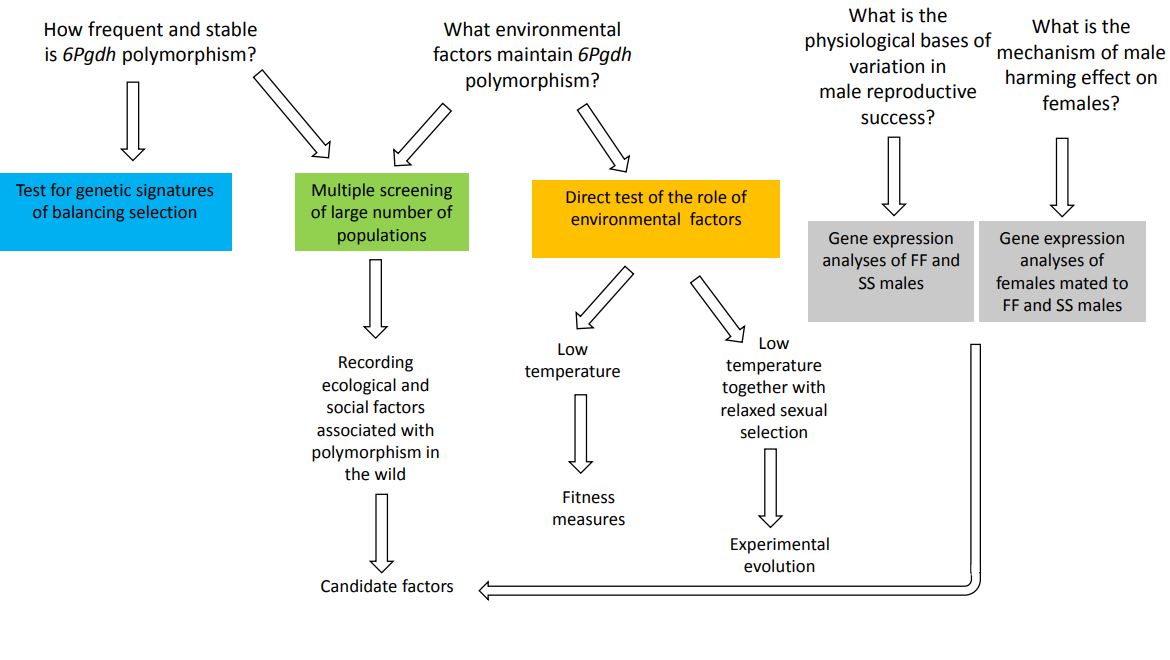
Zależny od środowiska dobór stabilizujący w genie związanym z konfliktem płciowym.


Tempo mutacji u kornika drukarza.

Rola pasożytów krwi w dynamice nowych chorób i utracie bioróżnorodności u płazów.

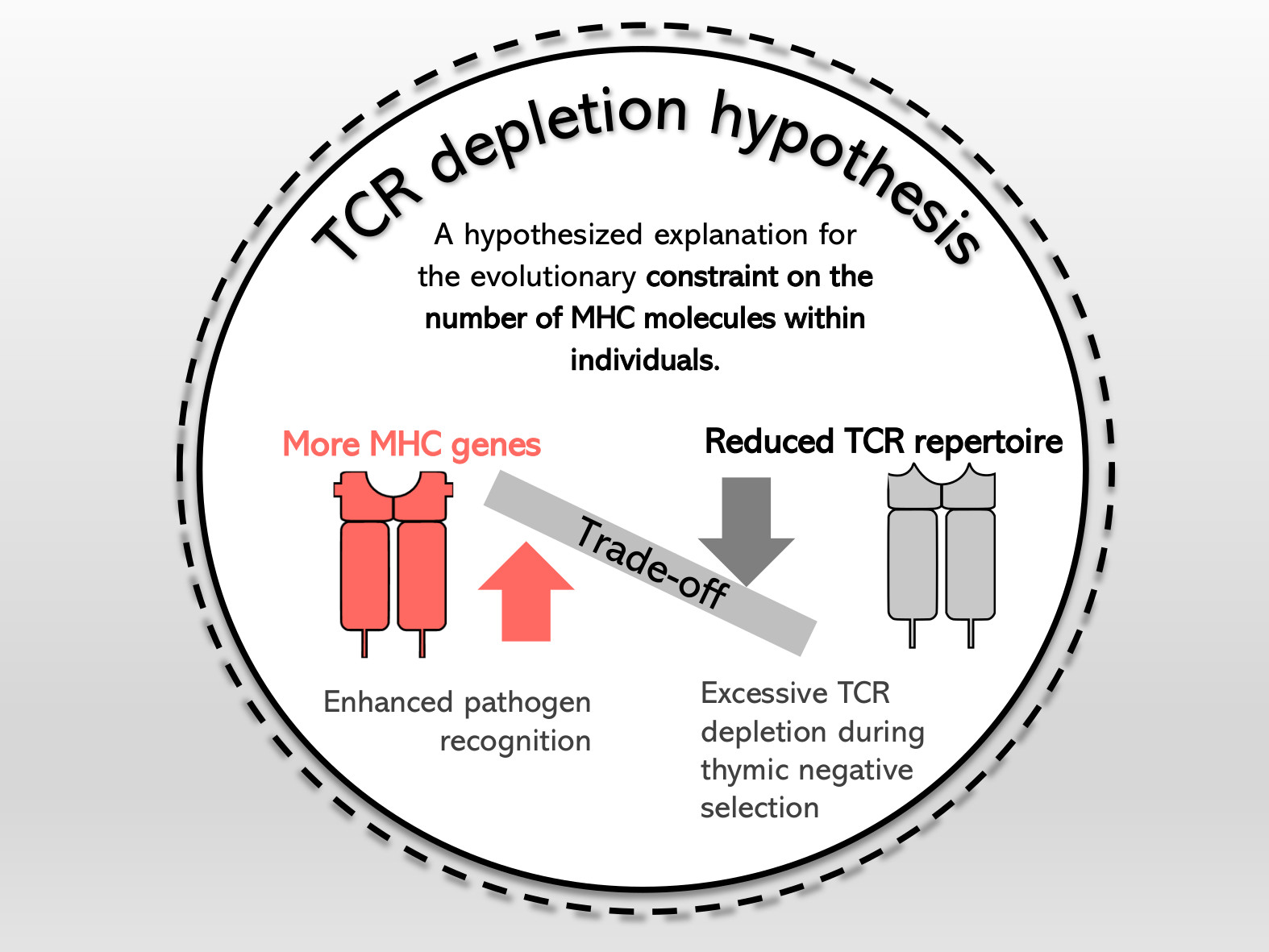
Ewolucja nieklasycznych genów MHC klasy I i repertuarów receptorów limfocytów T u salamander.